引 言
多器官功能障碍的发生是脓毒症中最主要的临床事件,因为其和患者的发病率和死亡率息息相关。尽管脓毒症的最新定义融入了这一概念,将脓毒症的临床本质集中在“由宿主对感染的反应失调引起的危及生命的器官功能障碍”的发生上,但我们对于脓毒症诱发器官功能障碍的机制的理解还很不全面。这种认知差距并非微不足道,是因为脓毒症的死亡率还是很高的,对于脓毒症患者的治疗选择非常有限且是非特异性的,此外脓毒症后其他疾病发病率对于出院患者也是很大的经济负担。
在过去的15年中,一些新的概念改变了我们对危重疾病背景下器官功能障碍的理解,并构建了可能导致器官功能障碍的机制研究。其中三个颠覆性想法在这里特别重要。首先,是在没有氧供减少的情况下,器官在脓毒症期间会出现功能障碍,这表明组织缺氧可能不是单独的机制。这就解释了为什么以灌注为目标的治疗努力可能只能获得部分益处或没有益处。第二,器官功能障碍可能在没有明显细胞死亡的情况下发生,表明功能障碍不是由于结构损伤,而是关闭了一些一般的细胞活动。这引发了人们的猜测,即器官功能障碍可能是一种应对炎症损伤的适应性策略。当然,如果这一过程持续下去,它将会导致适应失衡,并根据已知研究其与不良预后相关联。第三,是认识到免疫系统对入侵病原体的作用(也称为抵抗能力)只是身体抵抗感染的防御机制的一部分。直到最近才在植物生态学和生物学领域发现被称为免疫耐受的机制。这种机制在哺乳动物中有所描述,指的是宿主减轻免疫或病原体对细胞或组织所造成的伤害。通过实验研究得到的结论证明免疫耐受可以赋予器官保护和不依赖于宿主控制感染的能力(即抵抗力)的生存优势,这为研究器官功能障碍以及机体在病原刺激下的适应机制提供了框架。
重建组织灌注一直是感染性休克患者早期治疗抢救的基石。在多项研究中已观察到脓毒症患者存在微血管和内皮功能障碍、自主神经衰竭以及细胞内代谢调控。因此,许多研究人员建议针对这些机制中的一种或多种机制来减少脓毒症引起的器官功能障碍的发展。有趣的是,一些研究人员提到了免疫耐受的潜在作用(尽管会导致短暂的功能丧失),并推测这些“病理”改变中的一些(例如,生物能量调控)从长远来看可能会保护器官和组织。因此,本综述的目的是探讨目前对脓毒症中这些不同机制的理解及其与器官功能障碍的关系。我们的目标是探索其中的解释机制以及潜在的治疗靶点。
微血管功能障碍
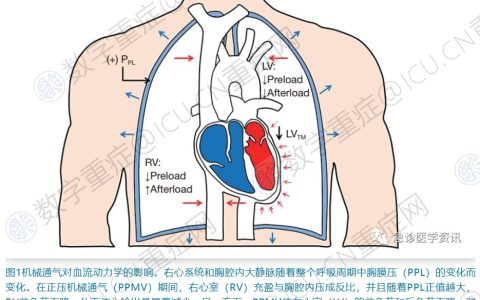
De Backer 及其同事的一项早期研究表明,通过手持正交偏振光谱成像技术监测舌下微循环,可发现脓毒症患者的微循环流量发生了改变。其特征性发现是灌注血管的比例减少,并且灌注不好(即间歇或停止流动)的血管比例增加,并且血供分布不均匀。这些发现现已被多项独立研究报告,并已在动物模型中的胃、小肠、结肠、肝脏和肾脏中得到证实。重要的是,舌下微循环流量的改变与脓毒症休克中器官衰竭和不良愈后息息相关。不太清楚的是,这些改变代表的是脓毒性相关性器官衰竭的因还是果。
线粒体功能障碍
尽管导致脓毒症微循环功能障碍的机制仍不完全清楚,但图 1 总结了当前的主要的框架机制。这些机制主要围绕于造成自我条件能力下降的机制,其中包括内皮细胞的损伤、逆行内皮细胞-细胞通讯改变、红细胞变形能力受损、血液粘度、糖萼剥落(一层有组织的糖胺聚糖层,覆盖内皮的管腔表面,具有关键的生物力学活性,包括维持血流和保护屏障功能)、血小板活化;白细胞粘附和滚动;和凝血和补体系统的激活。一氧化氮 (NO) 也可能导致微循环功能障碍。脓毒症的特点是整体 NO 产量增加;然而,NO 产生的关键催化剂之一,诱导型 NO 合酶 (iNOS) 的表达是不均匀分布的。这种分布可能与脓毒性微血管功能障碍的不均匀血流模式特征一致。值得注意的是,脓毒症中 iNOS 的选择性抑制可以改善肾脏微循环紊乱和减少肾功能损害。此外,具有抑制细胞聚集和促进局部血管舒张来保护内皮细胞作用的内皮 NO 合成酶衍生的 NO在脓毒症中减少,这也导致了微循环功能障碍。

内皮细胞/糖萼结构被破坏会导致间质水肿,这与脓毒症引起的器官衰竭直接相关。肾小球毛细血管内皮细胞糖萼似乎特别容易在脓毒症中降解。在一项盲肠结扎和穿刺 (CLP) 小鼠研究中,观察到尿白蛋白增加和肾小球滤过屏障的超微结构变化,同时 syndecan-1、透明质酸和唾液酸(所有重要的肾小球糖萼成分)的表达降低。这些屏障变化的原因与肿瘤坏死因子 (TNF)-α 有关,因为给予小鼠 TNF 导致的损伤类似于脂多糖 (LPS) 相关的肾小球超微结构变化,而LPS刺激没有TNF受体-1 的小鼠似乎不产生肾小球通透性的变化。在肺中观察到类似的糖萼破坏途径,表明存在共同的损伤途径。在肺中,肺泡内皮糖萼的破坏刺激内皮细胞液传导性增加,并导致肺水肿和脓毒症相关性肺病。在肝脏中,肝窦内皮结构改变与脓毒症相关肝细胞损伤和肝功能障碍相关。在小鼠 LPS 模型中,LPS 注射导致肝窦内皮的筛板结构丧失,导致流速降低,白细胞粘附和跨膜,并且血供的异质性增加从而导致供需不匹配。
微循环血供异常带来的后果
微循环血供异常会影响组织灌注。组织灌注取决于两个基本特征:扩散和传输。扩散取决于血管密度;随着血管密度的增加,氧气到达靶细胞所需的扩散距离会减少,反之亦然。传输取决于红细胞速度和血红蛋白饱和度。因此,毛细血管密度的降低(即毛细血管脱落)将通过扩散相关机制引起组织缺氧,而具有足够流量的血管比例下降(即,间歇性和停止流动的血管比例增加)将通过传输相关机制诱导组织缺氧。功能性毛细血管密度 (FCD),除以灌注血管密度 (PVD),反映了在给定区域内连续灌注的小血管(小于 20 μm)的数量。它整体评估了由于扩散(毛细血管密度)和传输(毛细血管血流)所造成的紊乱。从生理上讲,FCD/PVD 的减少会减少毛细血管交换的表面积,减少可用于扩散的氧气,并增加氧气和营养物质的扩散距离。
败血症中微循环改变的另一个特征是血流分布的异质性增加。尽管由于血流-代谢需求在健康个体中也会出现异质性,但脓毒症会导致血流异质性显着增加,可能会使氧气输送与需求脱钩,从而使氧气和营养物质的输送效率降低。图2描述了相对于正常或均匀减少的血供,增加的异质血供的血氧运输效率降低,特别是对于毛细血供血供超过需求的细胞(超量血供)相对于毛细血管血供和灌注血管太远的细胞(缺氧)。与代谢需求相匹配的灌注血管的毛细血管氧饱和度在脓毒症中较低,表明组织持续吸收输送的氧气。因为对氧气的提取取决于代谢需求,提供超量血供的毛细血管将有助于增加静脉氧饱和度 (SvO2)。观察到 SvO2 保持或升高时毛细血管氧张力的降低称为氧分压间隙。这表明局部氧气供应不足和存在功能性分流。血流不足以匹配局部组织代谢需求的特征还在于二氧化碳 (CO2) 清除减少,静脉 CO2 含量和分压增加(PCO2),导致 PCO2 的静脉动脉差异(PCO2 间隙)增加。在脓毒症期间,已记录到PCO2 间隙增加,并且已观察到 PCO2 间隙与微循环流量之间的反比关系。这表明存在缺乏CO2去除的停滞流动区域,或者换一句话讲存在组织缺氧的风险。此外,继发于内皮和糖萼损伤的脓毒症中的毛细血管渗漏制约了通过晶体液和胶体给药重建血管内容量,并具有促进间质水肿的作用。此外,间质水肿可能通过增加静脉压而影响血供,从而造成区域微血管瘀滞和PVD(舌下微循环)减少。

除了缺氧的风险外,局部血流速度降低可能造成炎症信号的放大有关。在内毒素血症的猪模型中,在心肌毛细血管中流动缓慢的区域观察到白细胞速度降低和白细胞转运时间增加。 由于延长转运潜在的伴随着暴露于分泌细胞因子的白细胞和病原体相关以及损伤相关的分子模式的时间增加,流动缓慢的区域显示出局灶性氧化应激和细胞损伤的迹象增加。在鼠 CLP 模型中,在病原体暴露 4 小时内观察到活性氧 (ROS) 和活性氮 (RNS) 增加,并且主要局限于血流减少或没有血流的区域。
代谢重组
抵抗和耐受
传统上,宿主对感染的反应归因于宿主免疫系统识别、靶向和消除外来微生物因子以控制病原体负荷的能力,这一过程称为抵抗。然而,病原体防御的另一种方法,首先在植物中描述,但后来在哺乳动物中描述,是宿主限制来自感染因子或其自身免疫反应的损伤的能力。这个过程被称为耐受,并且在特征上与抵抗无关。例如,在疟疾感染的情况下,血红素加氧酶 1 (HO-1) 的表达可以防止无细胞血红蛋白诱导的器官功能障碍,并已被证明可以提高生存率,这与宿主消灭载菌量的能力无关。尽管我们对宿主在应当不同危险时通过何种机制触发特定的免疫耐受所知甚少,但是这一概念提供了一个框架来理解代谢适应对细胞和器官保护的潜在作用,并描绘了其器官功能障碍方面的作用。
作为细胞生存策略的代谢重组
在早期脓毒症中观察到的细胞代谢下调,这种下调被认为是适应性的改变,其目的是重新确定能量消耗的优先级,以限制额外的损伤、维持能量平衡、防止 DNA 损伤和保持细胞成分。Hochachka 和 Guppy提出细胞位于持续缺氧环境的会减少对氧气的需求,这种机制称为氧气顺应性。Buck 和 Hochachka 证实了这一点,证明海龟肝细胞在缺氧状态下氧耗可以下调十倍,其主要通过分级关闭包括蛋白质合成在内的高能量消耗活动,同时保证维持生命必须的活动,例如保证细胞膜电位。Schumacker 及其同事在啮齿动物肝细胞中证明了类似的机制,在面对慢性(但非急性)缺氧,其呼吸频率抑制高达 40% 至 60%,证明这是跨物种的固有机制。
因此,短暂的功能丧失似乎是一种固有的细胞适应性机制,可以解释脓毒症引起的早期器官功能障碍。在心脏中,脓毒症会导致心肌细胞失去收缩性但不会死亡。这种下降与蛋白质下调和线粒体细胞色素 c 氧化酶活性降低有关。外源性细胞色素 c 可改善细胞色素 c 氧化酶活性并恢复心肌收缩力,这表明线粒体改变与器官功能丧失之间存在直接关系。在肺中,耗能过程,例如钠和氯转运蛋白以及与 ATP 酶连接的跨膜泵,在脓毒症期间被灭活和内化,一方面避免能量被过度使用,另一方面同时降低肺泡上皮清除肺泡内液体的能力并导致肺水肿(图 3)。在肾脏中,由LPS 或促炎细胞因子所诱导的炎症导致氯和钠通道的下调,这是肾小管上皮细胞中最主要的能量消耗(见图 3)。这种机制被认为将缺血再灌注中的肾小管损伤和肾小球滤过联系起来,因为近端小管以外的肾小管氯化物浓度(由于通道下调而不被重吸收)的增加会激活致密斑水平的肾小球反馈,导致通过收缩传入小动脉降低肾小球滤过率。然而,这种机制尚未在脓毒症的模型中得到证实。在肝脏中,在脓毒症中观察到排泄蛋白质的合成减少以及外源性和内源性毒素的转化受损。Hotchkiss 及其同事表明,人类脓毒症的特征是除肠道、免疫系统和脾脏外,其他器官中的细胞死亡很少,并且发现与其他器官相比,脾脏发生了更多的细胞凋亡。在脓毒症期间,由于上皮细胞凋亡增加和隐窝细胞增殖减少,肠道上皮细胞的屏障功能发生了改变,导致黏膜通透性增加和细菌易位。尽管能量调控在肠上皮细胞凋亡诱导中的作用尚不清楚,有证据表明 T CD4+ 淋巴细胞可以保护细胞减少凋亡;因此,脓毒症期间 T CD4+ 细胞凋亡可能导致肠上皮细胞凋亡。最后,应对脓毒症的细胞代谢重组可以通过几个协作的程序进行协调,包括代谢 ATP 生成的转变(在氧化磷酸化 [OXPHOS] 和糖酵解之间)、线粒体呼吸的抑制、清除损伤线粒体程序的激活,以及诱导细胞周期停滞。

代谢重组:从氧化磷酸化到有氧糖酵解
在即使有氧气(即有氧糖酵解),但通过糖酵解优先氧化葡萄糖的这种现象被称为 Warburg 效应其令人格外感兴趣。虽然其主要在癌细胞中观察到,但免疫细胞在炎症刺激时也使用这种机制。通过使用这种机制,免疫细胞改变能量产生的途径,从而通过氧化磷酸化维持管家功能,而激活所需的能量来自有氧糖酵解。此外,脓毒症的实验模型显示出在早期,大鼠CLP模型中的肾组织中代谢转变为有氧糖酵解。尽管能量效率低于氧化磷酸化,但使用糖酵解来给激活作用供能至少有两个优点。首先,它可以产生必需的结构成分,例如脂肪酸、氨基酸和核苷酸,同时产生足够的能量供细胞存活。其次,由于糖酵解中间体通过戊糖磷酸途径分流,增加的糖酵解导致烟酰胺腺嘌呤二核苷酸磷酸 (NADPH) 增加,这是减少线粒体 ROS 产生造成的氧化损伤的关键。另一方面,在脓毒症实验模型后期未能恢复氧化磷酸化已被证明会使促炎状态持续存在,从而影响器官功能和存活。因此,多项研究表明,在脓毒症实验模型中 氧化磷酸化启动子的激活对器官具有保护作用并可以提高存活率。炎症细胞中细胞代谢的变化已被证明是继发于重要细胞调节节点的复杂相互作用,其中包括Akt/哺乳动物靶向的雷帕霉素复合物 1 (mTORC1)/缺氧诱导因子 1 的α (HIF-1α) 通路初始促进转向糖酵解,和 sirtuins(尤其是 Sirt1 和 6)在后期促使转换回氧化磷酸化。

抑制呼吸电子传递链
脓毒症对线粒体呼吸和特定电子传递链复合物的影响已在动物模型和人类脓毒症中得到证实。在危重患者的骨骼肌活检中,线粒体电子传递链的复合物 I 和 IV 活性显着降低,其中非幸存者表现出更显着的活性降低和随之而来的组织 ATP 的更大下降。这些事件也已在大鼠脓毒症模型的肝脏中发现,表明这种现象不是骨骼肌特有的,而是一种潜在的普遍机制。线粒体复合物 I 和 IV 容易被亚硝基化的持续抑制,亚硝基化是一种由 RNS 易化的反应,在脓毒症的情况下其会升高,可以减少细胞死亡,并且重要的是,它是可逆的,从而可以恢复正常的器官功能。
线粒体细胞功能调节:线粒体自噬和生物合成
在脓毒症实验模型的早期阶段,在多个器官中观察到自噬增加,即细胞消化不必要或功能失调的成组分,这种现象已在人类脓毒症中得到证实。在 Toll 样受体 4(TLR -4) 介导的炎症、氧化应激、和电子传递链中 ATP 产生的非耦合呼吸所导致的线粒体膜去极化等病理刺激下会发生自噬。在这种情况下,脓毒症诱导的电子传递链复合物抑制可能作为一种信号机制激活自噬。自噬的激活具有临床意义,因为自噬反应的启动减少与疾病时间延长和器官功能障碍无法恢复有关。此外,通过用西罗莫司或表柔比星抑制 mTOR 来增强自噬可防止 AKI并改善生存率。同样在 LPS 诱导的小鼠脓毒症模型中应证这一结论。相比之下,通过阻断液泡分选蛋白 34 (VPS34)(一种促进自噬信号传导的中心蛋白)来抑制自噬,会导致肝功能障碍增加。自噬反应也被认为是炎症反应时代谢重组种的一个整体机制因为去除功能失调的线粒体和减少线粒体数量可以减少 ROS 诱导的损伤和 OXPHOS,特别是在向有氧糖酵解的急性转化中。
线粒体自噬通过氧化还原途径和依赖 TLR-9 的机制与线粒体生物合成相结合。线粒体池的补充可能是脓毒症恢复过程中不可或缺的一部分。因此,过氧化物酶体增殖物激活受体 γ 共激活因子 1-α (PGC-1α) 是线粒体生物合成的转录共激活因子,与病死者相比,在脓毒症幸存者的肌肉活检中显示其增加。此外,在脓毒症实验模型中吸入的一氧化碳即线粒体生物合成的外源性激活剂对肝损伤有保护作用。
细胞周期调控
线粒体也参与细胞周期的调节,并可能作为一种保护机制诱导细胞周期停滞。用最简单的术语来说,细胞周期是细胞通过特定阶段的进程,为细胞分裂做准备(包括有丝分裂的 G0、G1、S、G2 和 M)。在此过程中,细胞似乎使用特定的检查点来验证它是否已准备好进入下一阶段。从能量的角度来看,G1-S 检查点似乎很重要,因为正是在这个阶段,线粒体合并成一个巨大的网状结构,大概是为了增加 G2 期间 DNA 复制时的能量可获得性。在这些检查点期间,细胞似乎在验证它是否准备好进入下一阶段。如果没有准备好,细胞周期停滞可以防止复制的能量成本浪费,并且可以防止细胞损伤环境中的细胞死亡(图 5)。Yang 及其同事的研究结果支持这种预防措施,他们证明 G1-S 细胞周期停滞与 CLP 诱导的脓毒症后的 AKI 相关,并在恢复后进展至 G2。有趣的是,最近批准的 AKI 生物标志物、金属蛋白酶组织抑制剂 2 和胰岛素样生长因子结合蛋白 7 已知会诱导 G1 细胞周期停滞。这些生物标志物在识别包括脓毒症在内的危重疾病期间 AKI 发病风险优于其他标志物。

多器官交流
功能失调的器官可能会通过称为器官交流的复杂且不完全理解的生物通信过程影响其他远程器官。在 AKI 动物模型中观察到肾脑交流,其中继发于TNFα的局部肾脏炎症会造成被破坏的、更易渗透的血脑屏障和脑星形胶质细胞的激活。胶质纤维酸性蛋白,脑炎症的细胞标志物, 在AKI动物模型中升高,但在肝损伤动物模型中未升高,表明 AKI 具有特异性。在其他动物模型中,AKI 与脑神经递质浓度的改变和脑儿茶酚胺浓度的下降有关。尽管先前的神经功能正常,但肺损伤后患者会发生肺脑交流,随后发展为脑损伤。这种神经病理学在猪急性肺损伤模型中被证明与缺氧无关,在该模型中,通过肺灌洗和减少的吸入氧气分数实现了相似水平的低氧血症,最终用肺灌洗动物表现出更严重的脑损伤。
不可将交流与其他炎症综合征区分开来。例如,与高潮气量通气的大鼠相比,经 LPS 攻击的大鼠在中等呼气末正压(7 cm H2O)和低潮气量通气下表现出肺损伤和全身炎症减少;然而,在 LPS 和高潮气量组中,c-Fos(大脑中神经元激活的早期标志物)的表达在大脑的相同区域同样升高。已在临床前和临床研究中观察到,通过肾脏和肺之间的全身细胞因子的炎症互通与呼吸机诱发的肺损伤 (VILI) 相关高潮气量机械通气中 AKI 的发展有关。在肾-肺交流中似乎有一种独特的肾脏炎症模式。暴露于 CLP、VILI 和假手术的 AKI 小鼠模型显示,仅在 VILI 组的那些动物中,血管内皮生长因子(一种血管生成和内皮激活蛋白)和血管细胞粘附蛋白 1(一种白细胞粘附分子)升高,而在 AKI 和细菌性肺炎的小鼠模型中,单独的 AKI 不会引起临床上显着的急性肺损伤;然而,在细菌性肺炎的情况下,AKI 确实减弱了肺中性粒细胞的募集并增加了细菌载量,从而导致氧合受损。
虽然不是实体器官,但在脓毒症中,凝血和补体系统通过相互关联的途径相互影响。这种关系在早期脓毒症中观察到,同时出现强烈的补体激活和弥散性血管内凝血。补体终产物增加促进血液的血栓形成,诱导促凝和抗纤溶蛋白,并抑制抗凝途径。C3 转化酶抑制狒狒细菌性脓毒症可防止脓毒症引起的补体活化,减少血小板减少症和凝血功能障碍,并保留内皮抗凝特性。凝血因子也会影响补体激活。凝血酶和凝血因子 Xa 直接激活补体级联的组成成分;蛋白 C 抑制补体激活,凝血因子 XIIa 直接激活经典的补体途径。
自主神经系统
这种交流互通的概念扩展涉及在炎症环境中自主神经系统和免疫系统之间的交流。早期脓毒症中增加的循环中儿茶酚胺可增强初始炎症反应。除自主神经元外,白细胞还可以合成儿茶酚胺,并表达肾上腺素能受体,这表明神经递质会影响淋巴细胞运输、血管灌注以及免疫细胞增殖和凋亡。此外,门静脉引流的肠源性儿茶酚胺可以通过 α2-肾上腺素能受体信号传导改变肝枯否细胞和肝细胞状态。
由细胞因子和炎症分子刺激的迷走神经传入可促进迷走神经传出胆碱能信号传导,抑制 TNF 和其他促炎细胞因子的过度释放。这种胆碱能信号传导通过巨噬细胞和其他免疫细胞上的 α-7 烟碱乙酰胆碱受体传递。这种过度促炎信号所导致的炎症反射也延伸到脾神经与传出迷走神经通讯。在小鼠模型中,在 CLP 诱导的脓毒症后 24 小时刺激迷走神经可提高存活率。另一方面,迷走神经切断术与内毒素血症动物中促炎细胞因子水平升高有关。
小结
脓毒症是一种以显著临床异质性为特征的复杂综合征,其中发病率和死亡率和器官功能障碍息息相关。重要的是,脓毒症中的器官功能障碍现在被认为不仅仅是组织氧输送减少的结果,而是涉及对炎症的多种反应,包括内皮和微血管功能障碍、免疫和自主神经失调以及细胞代谢重组。实验数据表明,针对这些机制可能会导致器官保护并改善生存率。然而,它也强调,针对这些机制途径对短期和长期结果的影响在很大程度上取决于治疗干预的时机。在向前迈进时,努力了解这些机制的适应性或适应不良特征,发现特定阶段的生物标志物以指导治疗,并根据耐药性和耐受性概念化这些机制,将提供结构化的研究平台、转化途径和机会以改善患者的治疗效果。
本文荟萃自,只做学术交流学习使用,不做为临床指导,本文观点不代表数字重症 ICU.CN立场。
 微信扫一扫
微信扫一扫