脓毒症是一种严重损害健康的全身性疾病,由宿主对炎症反应的失调引起,是全球公共健康的优先事项[1]。尽管在临床治疗方面取得了许多进展,但脓毒症的发病率和病死率仍然很高[2],据研究统计表明,在全球范围内,脓毒症患者的重症监护病房(intensive care unit,ICU)病死率和住院总病死率分别为25.8%和35.3%[3]。在2016年,脓毒症被重新定义为由宿主对感染反应失调所导致的危及生命的器官功能障碍[4]。但对于脓毒症诱导的器官功能障碍的机制研究仍不清楚。近年来,人们越来越多地认识到铁代谢失调可能在脓毒症发病中起关键作用[5]。
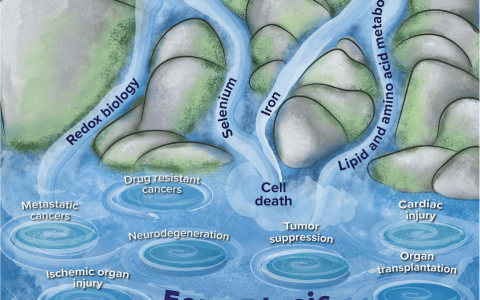
铁是生物体内完成许多生理基本过程所需的重要微量元素,包括蛋白质合成、氧代谢、细胞呼吸、DNA合成、脂质代谢以及免疫功能等[6]。2012年,Dixon等[7]首次提出了铁死亡的概念,这是一种依赖铁的、非凋亡的细胞死亡模式,其特征是脂质活性氧(reactive oxygen species,ROS)的累积。铁死亡作为一种新型的细胞程序性死亡方式,在形态和生化特征上不同于凋亡、自噬、坏死、裂解等多种形式的细胞死亡。从形态学特征上来看,主要表现为细胞膜断裂和出泡,线粒体变小,线粒体膜密度增大,线粒体嵴缩小甚至消失,线粒体外膜破裂,细胞核大小正常,染色质无凝聚[7,8]。从生化角度上来看,铁死亡的发生机制主要与谷胱甘肽(glutathione,GSH)耗竭、谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)失活、脂质过氧化和细胞内铁蓄积有关[9,10,11,12]。近年来的研究表明,铁死亡在脓毒症中的发生发展中起着重要的调节作用,已成为相关疾病诊疗和改善预后的研究重点和热点。因此,本综述将对铁死亡在脓毒症中的最新研究进展进行概述,以期进一步了解其发病机制并为脓毒症所致相关器官功能障碍提供新的治疗靶点。
1 铁死亡的机制
铁死亡的调节机制错综复杂,涉及多个代谢途径和信号通路,目前其分子机制的研究尚不清晰,仍处于不断完善的阶段。

1.1 铁死亡与氨基酸代谢异常
1.1.1 通过抑制胱氨酸-谷氨酸转运受体(XC-系统)诱导铁死亡:
GSH是铁死亡时氨基酸代谢的核心物质,由半胱氨酸、谷氨酸和甘氨酸合成[12],是细胞内主要的抗氧化剂之一,广泛分布于高等生物的组织中,可以保护细胞免受氧化损伤[13,14]。XC-系统是细胞内抗氧化系统的重要组成成分,其本质是一种氨基酸逆向转运体,是由SLC7A11和SLC3A2两个亚基组成的异二聚体,广泛分布于磷脂双分子层中。XC-系统可将细胞内的谷氨酸转运到细胞外间隙,同时将胱氨酸等比例转运到细胞内[7],被摄取的胱氨酸在细胞内被还原为半胱氨酸,而半胱氨酸可在谷氨酸-半胱氨酸连接酶和GSH合成酶的催化下生成GSH,从而进一步发挥其抗氧化作用[13,14]。因此,抑制XC-系统的活性,胱氨酸的吸收就会受到抑制,进而影响GSH的合成,使细胞的抗氧化能力下降,脂质ROS大量积累,最终发生氧化损伤和铁死亡。
此外,p53基因作为肿瘤中重要的抑癌基因,近年来也发现其可以通过下调SLC7A11的表达来特异性抑制XC-系统吸收胱氨酸,致使依赖胱氨酸的过氧化物酶活性降低,从而影响GPX4的活性,使细胞抗氧化能力下降,最终引起细胞铁死亡[15,16]。但p53具体通过何种机制调控铁死亡的发生仍需进一步研究。
1.1.2 通过直接抑制GPX4诱导铁死亡:
GPX4作为磷脂-过氧化氢GSH过氧化物酶的一种,其活性基团中含有一个硒代半胱氨酸,可提高其过氧化物酶的活性,降低脂质过氧化物的毒性,是铁死亡发生的关键调节因子[17,18]。GSH可以被GPX4转化为氧化型GSH,并提供电子将有毒的脂质氢过氧化物(L-OOH)还原为无毒的脂醇(L-OH)[19]。GPX4过表达可抑制ROS的产生和脂质过氧化[20],而GPX4活性或表达降低则使细胞内脂质过氧化物积累,最终导致铁死亡的发生[21]。以下是通过该途径发挥作用的几种常见的物质:①铁死亡诱导剂:Erastin通过影响GSH的合成降低GPX4反应底物的浓度,进而促进ROS的积累,间接诱导了铁死亡的发生[10,22]。GSH抑制剂RSL3则是通过直接与GPX4结合使其失活,抑制ROS的清除[23,24]。此外,RSL3还可以激活转铁蛋白(transferrin,TF),促使铁累积,最终通过促进过氧化反应加速ROS的产生[25]。②硒代半胱氨酸转运RNA(transfer RNA,tRNA):GPX4活性基团中的硒代半胱氨酸通过硒代半胱氨酸tRNA将其插入GPX4中[26]。而硒代半胱氨酸tRNA的成熟可通过甲羟戊酸(mevalonic acid,MVA)途径进行调节,从而影响GPX4的合成,间接调节铁死亡[27]。因此,通过抑制MVA途径下调硒代半胱氨酸tRNA的合成,也可以影响铁死亡的发生。③其他:细胞内的一些物质也可以影响GPX4的活性,如内过氧化物-1和2-二氧戊烷,可以间接使GPX4失活导致铁死亡[28]。铁死亡诱导剂FIN56可以通过降解GPX4,结合并激活角鲨烯合成酶(参与细胞内氧化水平的调节),进而导致脂质ROS增加,诱导铁死亡的发生[29]。因此,监测GPX4活性的降低将有助于观察铁死亡的发生情况。
1.2 铁死亡与脂质代谢异常:
脂质对维持细胞的形态和功能至关重要,包括生物膜组成、能量储存和信号传递。脂质代谢异常在铁死亡的发生发展中也起着重要的作用。
1.2.1 不饱和脂肪酸:
多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)是细胞膜脂质双分子层的重要组成部分[30],由于其分子结构中存在不稳定的碳-碳双键,因此对脂质的过氧化反应比较敏感[31]。有研究表明,铁死亡是由含有PUFAs的磷脂过度氧化引起的[32]。细胞内脂质过氧化的过程可大致分为非酶促脂质过氧化和酶促脂质过氧化两类。非酶促脂质过氧化是一种自由基介导的链式反应,而酶促脂质过氧化是指在脂氧合酶(lipoxygenase,LOX)的催化下产生各种脂质过氧化氢。非酶促脂质过氧化即脂质自氧化,本质是氧驱动的自由基连锁反应[33]。在细胞内有大量具有足够活性的自由基(如·OH)的情况下,脂质分子会泵出1个氢原子,产生脂质自由基,随后经泵氢、加成、断裂等不断重复的过程产生连锁反应。除非有足够数量的抗氧化剂来清除自由基,否则只要该反应占主导地位,氧化过程就不会停止[34]。芬顿反应就是如此,因此也被认为是脂质过氧化代谢的自由基提供者[35]。
目前,与铁死亡相关的脂质代谢的研究主要集中在酶催化的脂质过氧化反应上。已知的脂代谢中的重要酶有长链脂酰辅酶A合成酶4(long-chain fattyacyl-CoA synthetase 4,ACSL4)、溶血卵磷脂酰基转移酶3(lysophsphatidylcholine acyltransferase 3,LPCAT3)和花生四烯酸-15-脂加氧酶(arachidonate-15-lipoxygenase,ALOX15)[36]。花生四烯酸(arachidonic acid,AA)通常在ACSL4介导下先发生反应[37],然后在酰基辅酶A合成酶的催化下,产生AA-辅酶A(AA-coenzyme A,AA-CoA)和肾上腺酸-辅酶A(adrenic acid-coenzyme A,AdA-CoA),这两种PUFA-CoA的长链具有过氧化作用,能够破坏细胞膜。抑制ACSL4可以使AA-CoA和AdA-CoA的水平显著降低,干扰铁死亡的发生[32,38,39]。LPCAT3缺失可使膜中AA水平显著降低,导致血浆中甘油三酯蓄积和脂质过氧化,破坏细胞膜结构,进而导致铁死亡[40]。另外,ALOX15作为人类LOX的一种,也参与细胞铁死亡的发生过程,其过表达可增加相关氧化性PUFAs的浓度,最终导致铁死亡的发生[41,42]。因此,抑制脂代谢过程中相关酶的表达可以减少细胞内脂质过氧化物的积累,从而抑制铁死亡。
1.2.2 脂质ROS:
铁死亡从本质上来讲是脂质过氧化造成的细胞损伤,细胞内抗氧化功能被抑制或丧失可诱导此过程的发生[7]。已有的研究表明,细胞内脂质ROS主要通过以下几个途径产生:①哈伯-毛斯(Haber-Weiss)反应[43]和铁介导的芬顿反应可产生ROS[35]。②通过LOXs催化不饱和脂肪酸发生氧化,形成有毒的脂质过氧化产物,参与脂质ROS的生成[44,45]。当细胞质中存在大量铁离子时,可产生自由基使邻近PUFAs的质子发生转移,从而启动新一轮的脂质氧化反应,进一步传递氧化性损害[31]。③磷酸戊糖途径(pentose phosphate pathway,PPP)是产生还原型烟酰胺腺嘌呤二核苷酸磷酸(reduced nicotinamide adenine dinucleotide phosphate,NADPH)的主要来源。NADPH作为细胞内重要的还原剂,可以通过消除脂质过氧化来减少铁死亡的发生[46,47]。因此,抑制相关反应的酶或直接干预PPP可以改变细胞抗氧化能力和对铁死亡的抵抗。
通过影响以上几个反应途径可以干预铁死亡的发生,同样,使用相关抗氧化剂或抑制氧化反应过程也可以减轻铁死亡。①抗氧化剂:维生素E作为一种脂溶性抗氧化剂和铁螯合剂,可以降低细胞膜的氧化水平,抑制由GPX4减少而引起的铁死亡[48,49]。此外,由MVA途径产生的辅酶Q10(coenzyme Q10,CoQ10)是一种内源性抗氧化剂。有研究表明,铁死亡抑制蛋白1(ferroptpsis suppressor protein 1,FSP1)是CoQ10的一种氧化还原酶,可以在质膜上还原CoQ10,从而减轻铁死亡对细胞的损伤[50]。②分子调控:GPX4 mRNA的表达需要整合素α6β4参与。整合素α6β4缺失可以使GPX4的水平降低,间接导致脂质过氧化和铁死亡[51]。另外,有研究表明,热休克蛋白A5(heat shock protein A5,HSPA5)已被证明可以稳定结合GPX4,提高细胞的抗氧化能力,负性调节铁死亡[52]。③其他:半胱氨酸脱硫酶(cysteine desulfurase,NFS1)可介导细胞内铁硫簇的产生,而铁硫簇作为蛋白质辅因子,可维持细胞氧化水平,减少ROS的积累[53,54,55]。因此,铁硫簇生成障碍也会影响细胞铁死亡。
1.3 铁死亡与铁代谢异常:
铁是依赖铁催化的各种生物功能所必需的微量元素[56]。铁代谢在铁死亡过程中扮演着重要的角色。正常情况下,由小肠吸收或红细胞降解形成的二价铁离子(Fe2+)被氧化成三价铁离子(Fe3+),然后与细胞膜上的TF结合形成TF-Fe3+,之后通过与膜上的转铁蛋白受体1(transferrin receptor-1,TFR-1)结合以内化的方式进入细胞[57]。进入细胞后,Fe3+可转化为铁蛋白储存起来,也可被前列腺跨膜上皮3抗原(six-transmembrane epithelial antigen of protein 3,STEAP3)还原为Fe2+,然后Fe2+通过二价金属转运蛋白1(divalent metal transporter 1,DMT1)或锌转运蛋白8/14释放到细胞不稳定铁池中[57]。正常情况下,细胞内的铁处于动态平衡,当这些蛋白的功能出现障碍或表达异常时,将会导致细胞内铁离子浓度失衡,进而导致铁超载。当细胞中存在过量的铁时,便会通过芬顿反应直接催化脂质ROS的产生,最终引发铁死亡[10]。
近年来的研究也发现了一些通过调节铁代谢进而影响铁死亡的途径。①编码TFR-1的TFRC基因沉默可以抑制铁死亡诱导剂Erastin诱导的铁死亡[58]。②HSPβ1也可以通过抑制TRF-1的表达降低细胞内铁的浓度,进而影响铁死亡[59]。③血红素加氧酶1(hemo oxygenase-1,HO-1)是一种可以催化血红素生成亚铁、一氧化碳和胆绿素的胞内酶。HO-1上调可以发挥抗氧化作用,保护细胞免受ROS攻击。但过度激活HO-1可导致大量Fe2+在细胞质中积聚,促进ROS的产生,最终导致铁死亡[60]。④核因子E2相关因子2(nuclear factor-erythroid 2-erythroid 2-related factor 2,Nrf2)是调控抗氧化应激的一种关键转录因子,激活Nrf2可以减少铁的吸收,抑制ROS的产生,增强细胞的抗氧化能力[61]。⑤铁蛋白由铁蛋白轻链(ferritin light chain,FTL)和铁蛋白重链1(ferritin heavy chain 1,FTH1)组成,是细胞内储存铁的主要形式。铁反应元件结合蛋白2(iron responsive element binding protein 2,IREB2)是铁代谢的主要转录因子,抑制其表达可以显著增加FTL和FTH1的表达,从而抑制铁死亡的发生[62]。⑥核受体共激活因子4(nuclear receptor coactivator 4,NCOA4)可以特异性地识别铁蛋白并将其转运到溶酶体,随后铁蛋白中的铁离子被溶酶体释放到胞质中,产生大量的自由基,通过芬顿反应直接造成细胞的氧化损伤[63,64]。研究显示,NCOA4可通过激活ATG5-ATG7-NCOA4自噬通路促进铁死亡,因此,下调NCOA4的表达或沉默相关自噬基因(如ATG3、ATG5、ATG7和ATG13),可减少铁蛋白降解和铁的累积,抑制铁死亡[65]。
2 铁死亡与脓毒症诱导的多器官功能障碍
近年来的一些研究表明,铁死亡与脓毒症导致的多器官功能障碍密切相关[66]。
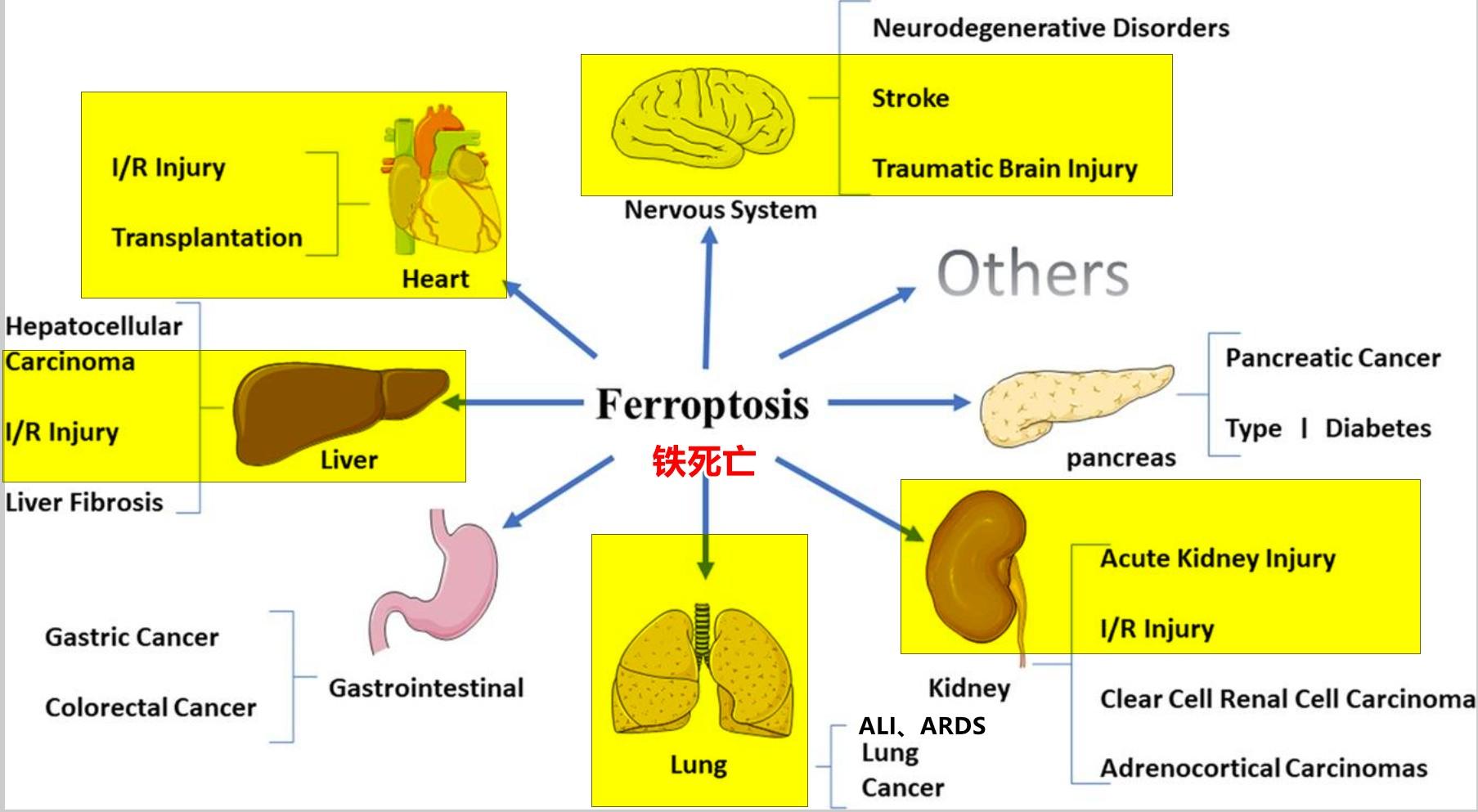
2.1 铁死亡与脓毒性心肌病:
脓毒性心肌病是脓毒症所致的急性心功能障碍,是一种可逆的并发症,也是脓毒症高病死率的主要原因之一,主要表现为左心室扩张、心室收缩功能和(或)舒张功能被抑制,导致射血分数降低和心源性休克[67]。合并脓毒性心肌病的患者病死率会增加65%[68]。既往研究表明,用脂多糖(lipopolysaccharide,LPS)诱导的脓毒性心肌病常引起心肌细胞凋亡、坏死以及铁死亡等,与脓毒症所致心功能障碍密切相关[69]。此外,在脓毒症小鼠模型的心脏中也发现铁死亡标志物之一环氧合酶-2 (cyclooxygenase-2,COX-2),即前列腺素内过氧化物合酶2的高表达[70]。而且还发现LPS诱导的心肌线粒体损伤的形态学变化与铁死亡的线粒体变化特征一致[71]。进一步研究显示,在LPS诱导的脓毒性心肌病模型中,LPS激活NCOA4,使其表达增加,NCOA4可特异性识别并降解铁蛋白,释放出大量的Fe2+,导致铁蓄积,最终引发铁死亡[72]。此外,用铁死亡抑制剂铁抑素(Ferrostatin-1)和铁离子螯合剂可显著降低脓毒性心肌病动物的死亡率,而铁死亡诱导剂Erastin和Sorafenib则通过耗竭GSH加重LPS诱导的细胞损伤[73]。综上所述,铁死亡与LPS诱导的脓毒性心肌病关系密切,针对心肌细胞铁死亡的措施可能是治疗脓毒症所致心脏损伤的一种新靶点。
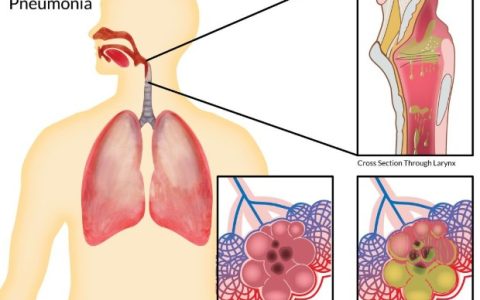
2.2 铁死亡与脓毒症诱导的急性肺损伤(acute lung injury,ALI):
脓毒症导致的肺损伤本质上是由于机体对炎症反应失控而引起的ALI,通常发生在脓毒症的早期阶段[74]。近年来,大量的研究表明,铁死亡与ALI的发生相关。铁死亡时ROS诱导的氧化损伤和炎症活动进一步促进炎症因子释放,加重ALI的发生[75],ALI时的炎症失控又进一步促进了ROS在肺组织中的积聚,促进细胞发生铁死亡[76,77]。有研究表明,脓毒症时肺泡上皮细胞铁水平升高,促进脂质发生过氧化[78]。此外,在LPS诱导的ALI动物模型中发现,总铁含量、丙二醛(malondialdehyde,MDA)和4-羟基壬烯醛(4-hydroxynonenal,4-HNE)水平显著升高,而SLC7A11和GPX4的表达明显降低[76]。另外,有研究显示,Nrf2可以通过减少GSH消耗,增加GPX4的表达和线粒体功能,减轻铁死亡和肺损伤[79]。使用铁死亡抑制剂铁抑素-1也可以抑制ALI的进一步加重[76]。近年来,在抑制铁死亡和减轻ALI的信号通路方面研究较为广泛,尤其是Nrf2/缺氧诱导因子-1α(hypoxia-inducible factor-1α,HIF-1α)和Nrf2/HO-1/SLC7A11通路[80]。有研究用衣康酸(Itaconate)治疗LPS诱导的小鼠ALI,结果显示,衣康酸可通过上调Nrf2抑制巨噬细胞的铁死亡,减轻脓毒症引起的ALI的病理改变[81]。这些研究初步证实了铁死亡可能是脓毒症所致肺损伤的潜在治疗靶点,也为临床治疗ALI提供了新的思路。
2.3 铁死亡与脓毒症诱导的急性肾损伤(acute kidney injury,AKI):
脓毒症所致的AKI又称为脓毒症相关性AKI(associated-AKI,SA-AKI),使严重脓毒症患者的住院时间和住院病死率显著增加[82],在已经住院的危重患者中,尤其是ICU患者,AKI发生的主要原因是脓毒症[83]。
先前的研究表明,在SA-AKI中发现肾小管细胞坏死、凋亡和自噬[84,85,86,87,88]。近年来的一些研究揭示了铁死亡在SA-AKI中的作用。研究显示,GPX4基因敲除小鼠AKI的发生率和死亡率显著增加,清除其体内脂质过氧化物可提高约35%的存活率[89]。上调SA-AKI小鼠肾脏中Nrf2的表达,可有效抑制脓毒症小鼠铁死亡和AKI的发生[90],使用铁抑素-1也可显著减轻SA-AKI[91]。另外,有研究也显示,使用铁螯合剂可减轻Erastin和RSL3诱导的近端肾小管上皮细胞的氧化应激和死亡[92]。除此之外,由于脂质过氧化会导致血管强烈收缩和氧化损伤,是AKI发生的不利因素[93]。有研究也表明,早期SA-AKI患者血清中LPCAT3活性升高[94],肾脏组织MDA和4-HNE含量在LPS诱导的脓毒症小鼠中显著增加[95],而去铁胺可以显著抑制脂质过氧化和肾小管上皮细胞的坏死,预防肾功能衰竭[96],减少铁超载,从而缓解脂质过氧化造成的细胞损伤[97]。综上所述,铁死亡在SA-AKI的发生中也起着重要的作用,通过抑制铁死亡的发生可有效提高SA-AKI动物模型的治疗效果。
3 总结和展望
综上所述,铁死亡作为一种新的细胞程序性死亡方式,引起了越来越多的关注。随着对其研究的不断深入,也发现铁死亡参与脓毒症相关器官功能障碍的发生发展。但目前对于铁死亡的研究还有许多问题有待进一步考证。除了目前所熟知的几种铁死亡调节途径之外,是否还有其他调控机制参与尚不清楚,且与其他细胞死亡方式相比,铁死亡缺乏相应的特异性标志物,因此,对于铁死亡的特异性标志物的研究具有重要意义。除此之外,脓毒症的发生发展过程也伴随着细胞的铁死亡,抑制铁死亡使脓毒症相关器官功能障碍的预后得到改善,有较好的研究前景,对铁死亡抑制剂的作用机制以及作用靶点的研究仍需要进一步探索。虽然在动物和细胞实验方面已经证实铁死亡与脓毒症相关器官功能障碍之间存在关系,但如何将这些研究发现应用于脓毒症相关器官功能障碍的临床治疗也是目前亟待解决的问题。总之,在脓毒症状态下对铁死亡的探索和认识仍处于起步阶段,深刻认识铁死亡在脓毒症中的调控机制以及针对性的临床干预将有助于更精确的诊疗和防治。
来源:中华危重病急救医学, 2022,34(9) : 985-990.
本文荟萃自公众号,只做学术交流学习使用,不做为临床指导,本文观点不代表数字重症 ICU.CN立场。
 微信扫一扫
微信扫一扫